
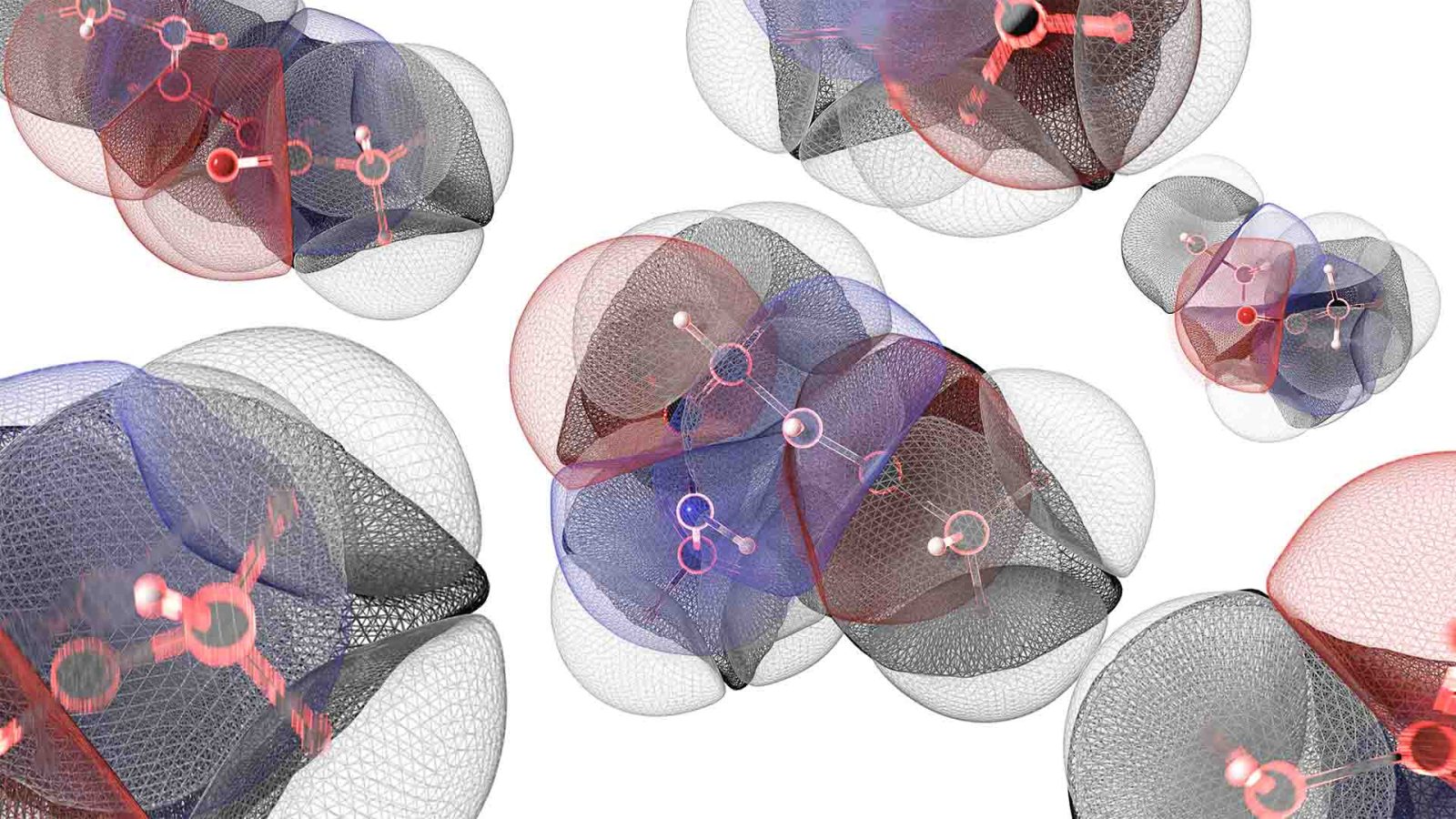
The study, published in Communications Chemistry, explores the first AI‑powered model that can keep molecular simulations running safely and smoothly, even when molecules are pushed to extreme conditions. In simple terms, this model stops molecules from “breaking apart” inside the simulation, allowing researchers to study how they behave over long periods and at very high temperatures. This stability opens the door to more reliable discoveries in areas like drug development, new materials and sustainable chemistry, all without relying on expensive supercomputers.
Building more reliable AI molecular models
Machine‑learned potentials (MLPs) are widely used to approximate quantum mechanical behaviour in molecules, but most existing models become unstable when molecules experience heat, movement or structural distortion. This makes long, reliable simulations extremely difficult to achieve.
The Manchester team – Bienfait Kabuyaya Isamura, Olivia Aten, Mohamadhosein Nosratjoo and Professor Paul Popelier – has solved this long‑standing challenge by integrating deep physical knowledge directly into their model.
The researchers built a new AI model using Gaussian process regression, to understand how atoms in a molecule naturally behave. To do this, they fed the model detailed information about how atoms interact in real life, based on the rules of quantum physics, to help the AI make more realistic predictions about how each part of a molecule should move.
They also discovered that a small mathematical choice, called the “prior mean function”, affected the stability of the model; with this function in place, the AI had the correct “starting point” to create and sustain a stable model even when a molecule is stretched, heated or shaken.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave a comment